Molecular replacement with MRage
This tutorial explains how to use to run phaser.MRage from the Phenix GUI (input files to run the same through the command line examples are also available).
Beta/blip
This is a complex between beta lactamase (beta) and beta lactamase inhibitor (blip). There are good models available, but the space group is ambiguous. This tutorial shows basic MRage functionality with a two-component system.
Experimental data for this tutorial is distributed with Phenix GUI and is also available from the Tutorials page.
Input
The MRage GUI is available from the main Phenix window under the Molecular replacement tab (select the Enable alpha features options under File/Preferences dialog for it to show up). On startup, the GUI presents the Basic options dialog.
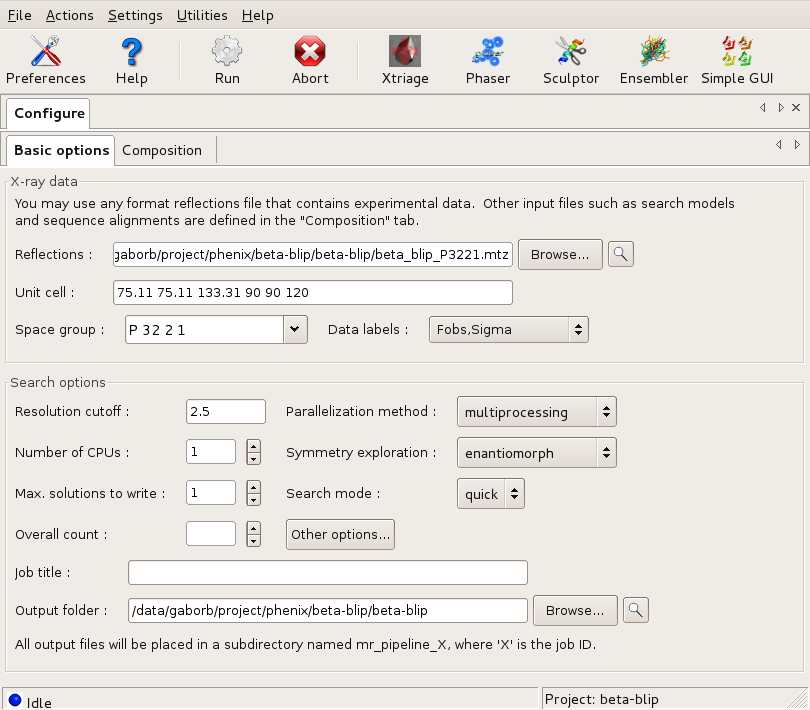
The following screenshot shows the this required settings to run the tutorial. Note, that almost everything is filled in automatically once the reflection file has been selected, or default. The only additional change one needs to make is to switch the space group exploration box to enantiomorph from its default value dataset, as the space group is enantiomorphic, and it is not possible to know without solving the structure, which hand is the correct one. In this tutorial, only a single CPU is used, but feel free to use more if available! Note that the Overall count box has been left blank. This tells the program to determine the number of copies automatically.
 The Basic options dialog Large |
{kind=link}
Next, we need to tell the program about the composition of the asymmetric unit and the available search models.
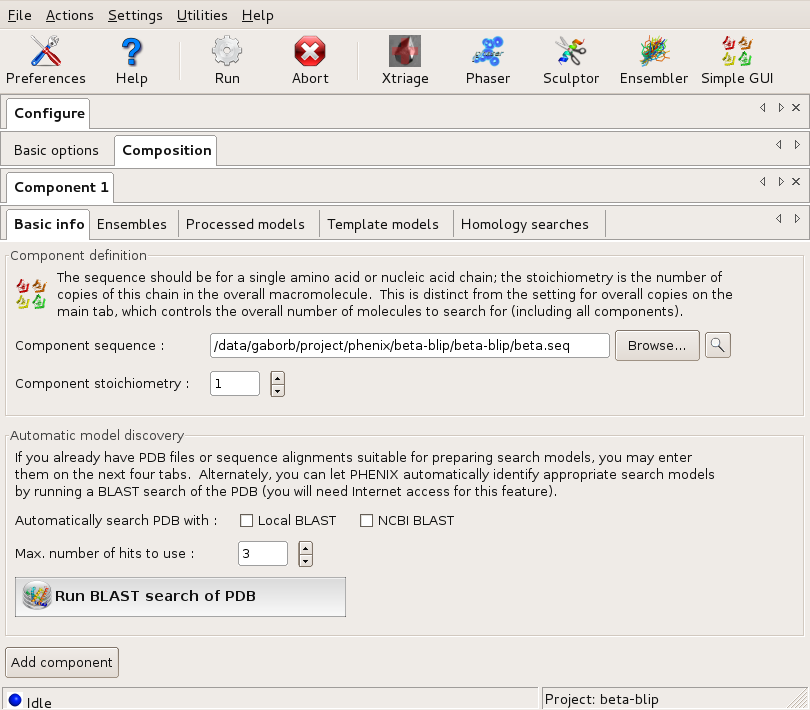
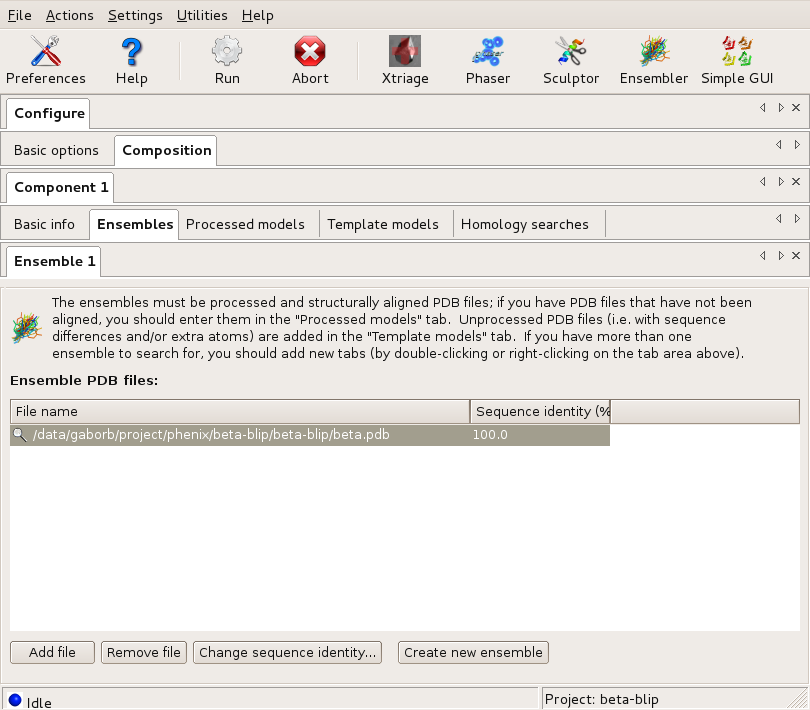
First, the beta component is described with its sequence, and the only search model. Since there is no modification needed to this model (it is 100% identical), it is input through the Ensemble tab. This tells the program that there is one alternative model for the beta component.
 The Basic info dialog of Component1 (beta) Large |
 The Ensembles dialog of Component1 (beta) Large |
{kind=link}
{kind=link}
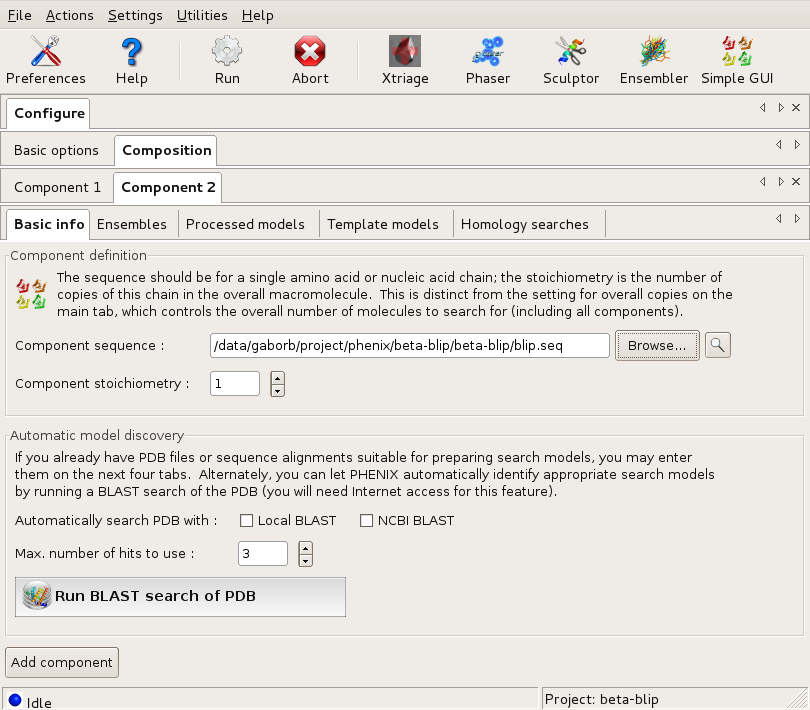
Then, a new component is created for blip (using the Add component button on the Basic info tab). This is filled out accordingly.
 The Basic info dialog of Component2 (blip) Large |
 The Ensembles dialog of Component2 (blip) Large |
{kind=link}
{kind=link}
The job is ready to run!
- Running from the command line
Input file (betablip.eff)
hklin = beta_blip.mtz labin = Fobs,Sigma symmetry_exploration = enantiomorph
composition
{
component
{
sequence = beta.seq
ensemble
{
coordinates
{
pdb = beta.pdb
identity = 1.00
}
}
}
component
{
sequence = blip.seq
ensemble
{
coordinates
{
pdb = blip.pdb
identity = 1.00
}
}
}
}
Command line
phaser.MRage --verbosity=VERBOSE betablip.eff
Output
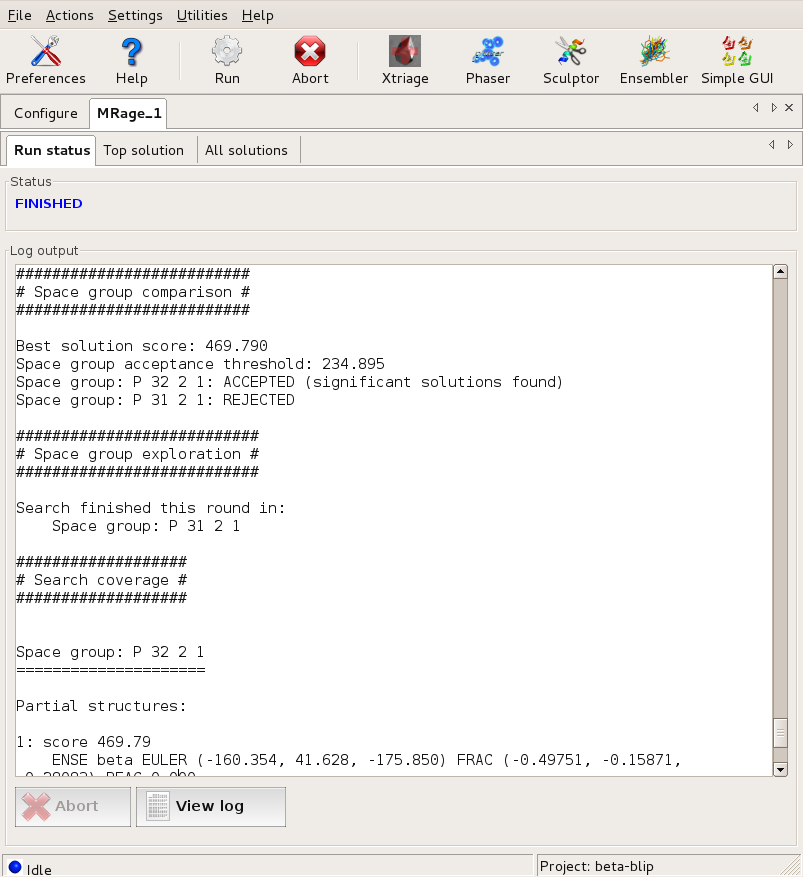
The GUI displays the program output in the logfile window. First, the program starts searching in both possible space groups.
 After finding the first component, the search in P3₁21 is dropped... Large |
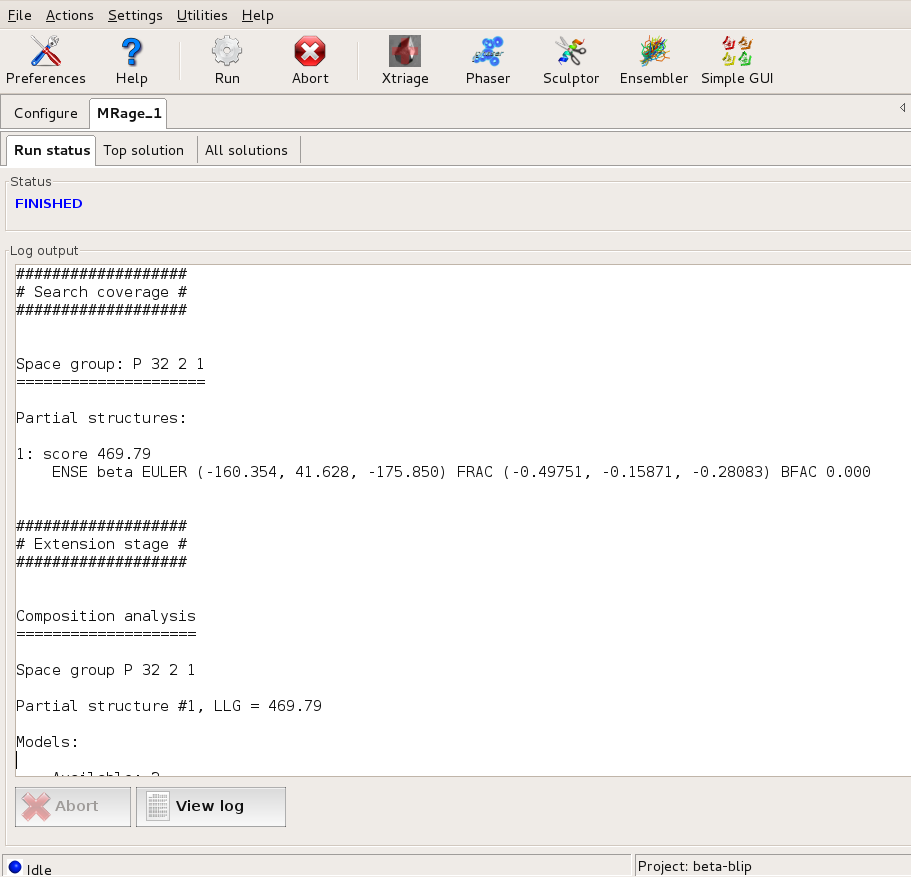
 ...and only P3₂21 is propagated with the partial solutions found. Large |
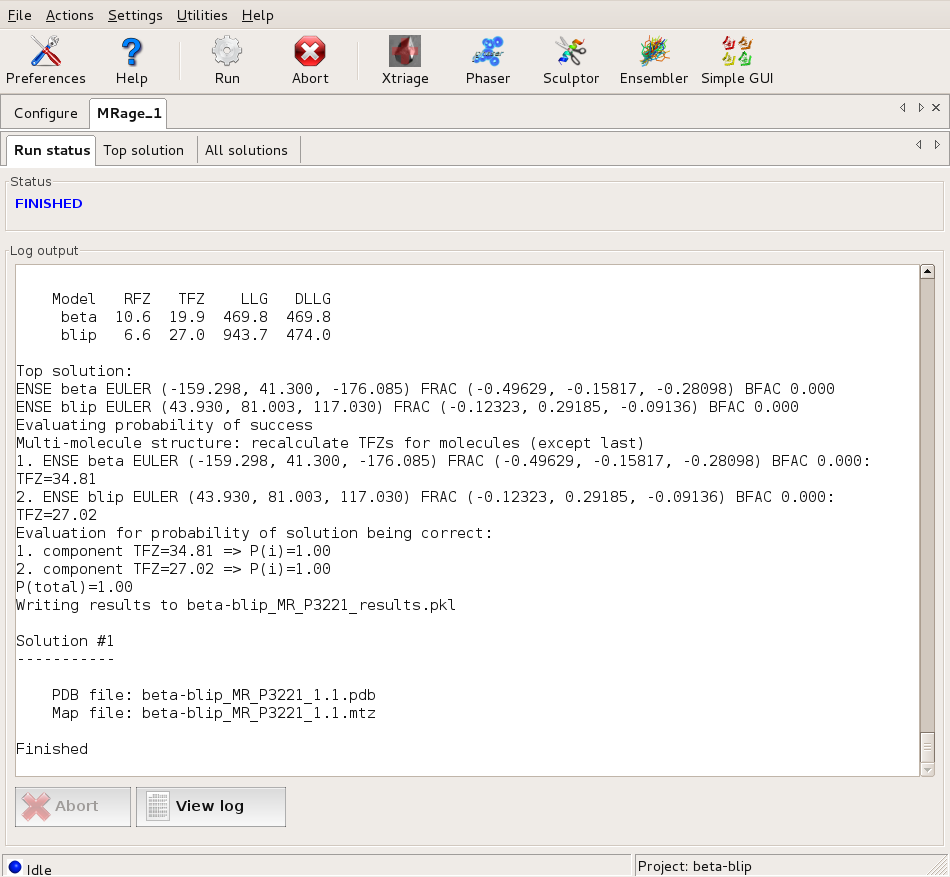
 The final solution: evaluation shows a high probability for it to be correct. Large |
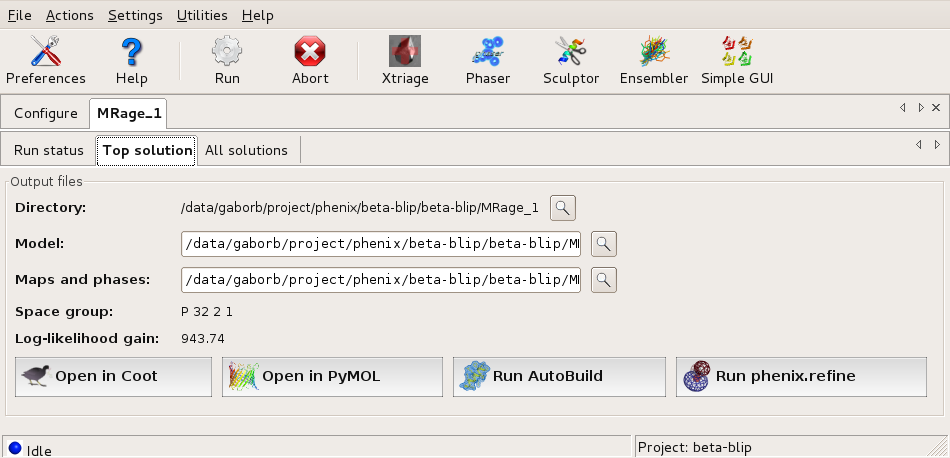
 Final solution shown in the GUI Top solution tab with possible further steps. Large |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lysozyme
As a frequently used test protein, there are good models for lysozyme. This tutorial demonstrates how to solve lysozyme starting from a homology search.
Input
 Basic options dialog. Note that the space group is enantiomorphic, but we pretend we know the correct space group P4₃2₁2 to save time |
 Setting up composition. It would be possible to tick the NCBI Blast option and run, but we want to avoid overloading the NCBI and provide a saved homology search file |
 Specifying the homology search file. There are many hits, and the GUI by default selects the first three. Selecting any larger number, however, makes no difference! |
- Running from the command line
Input file (lysozyme.eff)
hklin = lyso.mtz labin = "F_New,SIGF_New,DANO_New,SIGDANO_New,ISYM_New"
composition
{
component
{
sequence = hewl.seq
homology
{
file_name = hewl_ebi_wu_blast.xml
max_hits = 3
}
}
}
Command line
phaser.MRage --verbosity=VERBOSE lysozyme.eff
Output
 The program fetches the first homologue... |
 ...then processes it with sculptor. Note that although all 13 protocols in sculptor are active, it only creates 7 models, because remaining protocols create models that are identical to the ones used. |
 This model gives a clear solution, and processing stops. Although 3 hits were specified, the second and the third was never downloaded, because the first gave a clear solution |